

Precision and effective applications for above and below the knee.
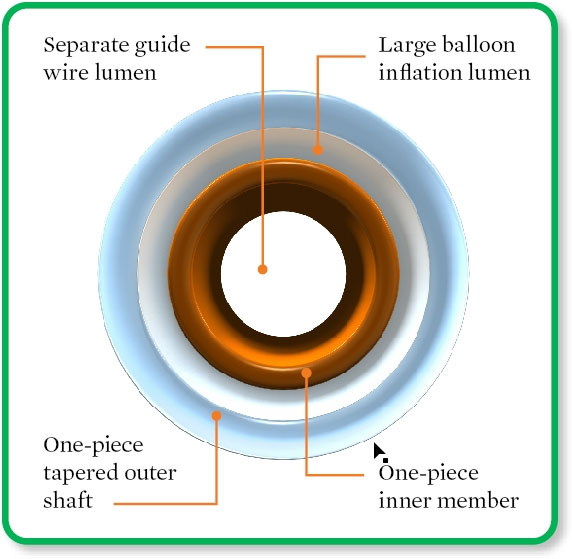
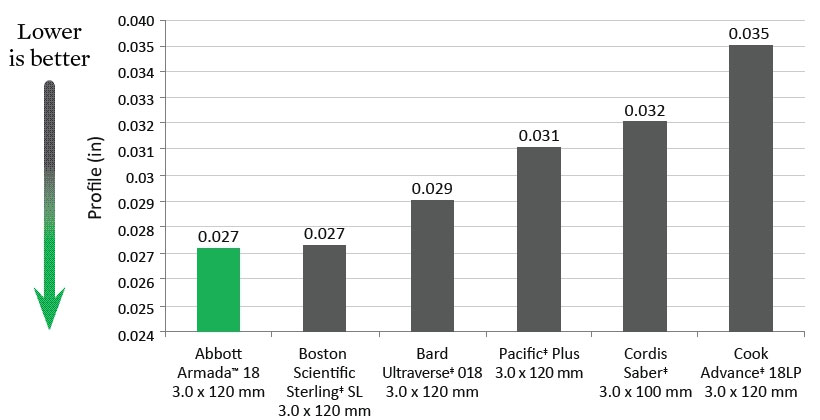
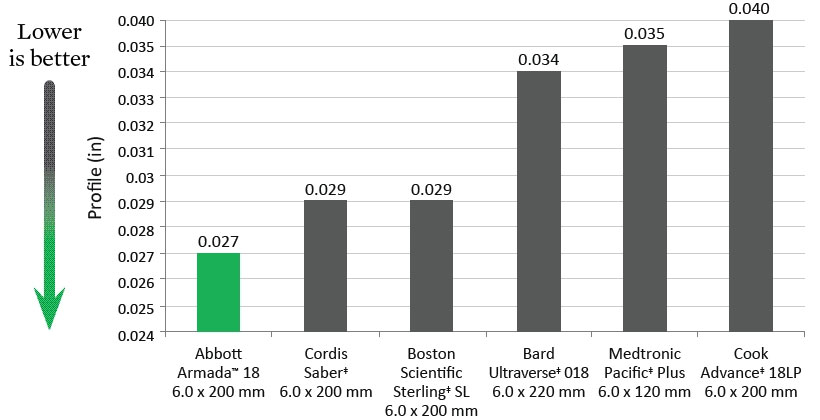
SFA Tip Entry (1 mm from tip)
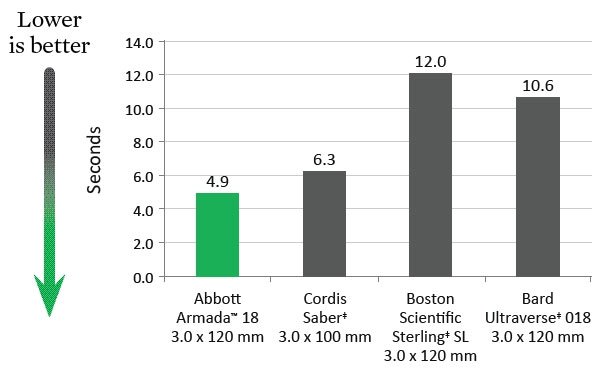
Deflation Time

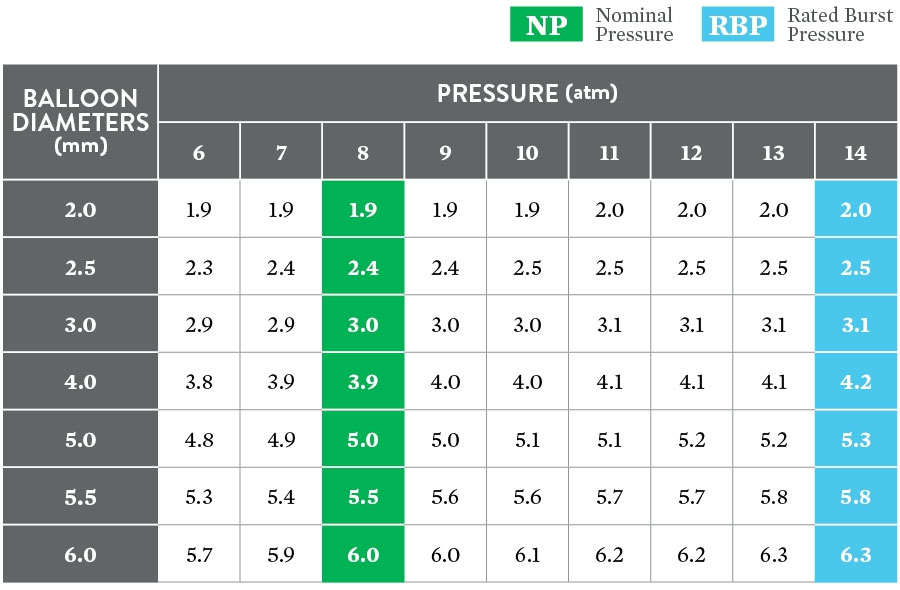
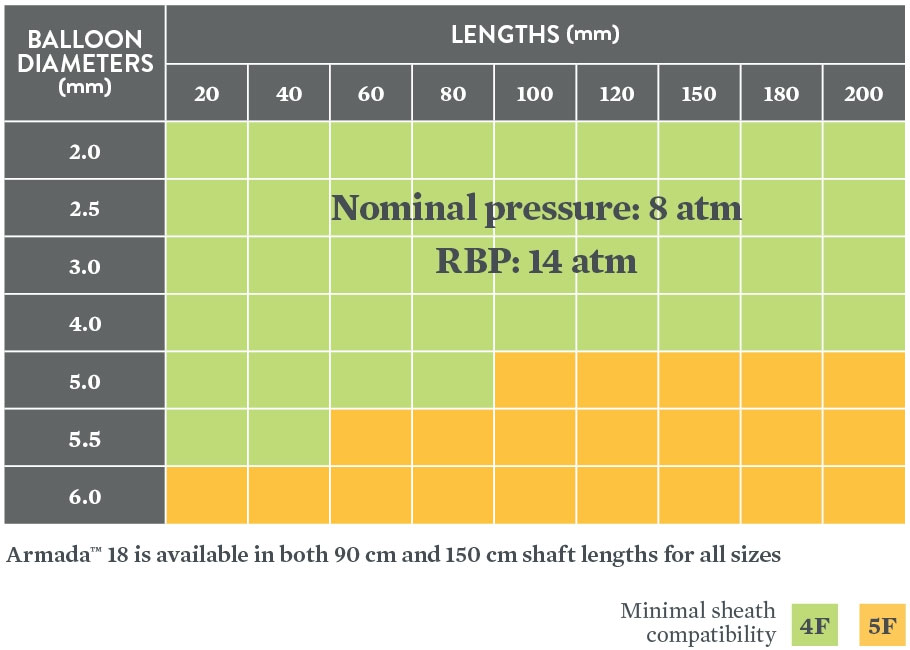
Data on file at Abbott.

MAT-2114521 v1.0
The Armada™ 18 is indicated to dilate stenosis in femoral, popliteal, infra-popliteal, and renal arteries and for the treatment of obstructive lesions of native or synthetic arteriovenous dialysis fistulae. In addition, the device is also indicated for post-dilatation of balloon expandable and self-expanding stents.
The following complications may occur as a result of PTA, but may not be limited to:
MAT-2114523 v1.0
Indications
The Supera™ Peripheral Stent System is indicated to improve luminal diameter in the treatment of patients with symptomatic de novo or restenotic native lesions or occlusions of the superficial femoral artery (SFA) and / or proximal popliteal artery with reference vessel diameters of 4.0 to 7.5 mm, and lesion lengths up to 140 mm.
Contraindications
The Supera™ Peripheral Stent System is contraindicated in:
Warnings
Precautions
The Supera™ Peripheral Stent System should only be used by physicians and medical personnel trained in vascular interventional techniques and trained on the use of this device.
Magnetic Resonance Imaging (MRI) Safety Information
Nonclinical testing has demonstrated that the Supera™ stent, in single and in overlapped configurations up to 250 mm in length, is MR Conditional. A patient with this device can be safely scanned in an MR system meeting the following conditions:
Under the scan conditions defined above, the Supera™ stent is expected to produce a maximum temperature rise of 7.6 °C after 15 minutes of continuous scanning.
In nonclinical testing, the image artifact caused by the device extends approximately 2 cm from the Supera™ stent when imaged with a gradient echo or spin echo sequence and a 3T MRI system.
Potential Adverse Events
Potential adverse events include, but are not limited to:
MAT-2103597 v3.0
STAY CONNECTED